
Table of Contents
What is Parkinson’s Disease?
Parkinson’s disease is a relatively well-known disease that can significantly influence the ability to function. Therefore, it’s likely that many of us want to know what Parkinson’s disease is.
The answer to this question began to be discovered in 1817, when James Parkinson published a text called, “Essay on the Shaking Palsy”. This book described the basic symptoms of Parkinson’s disease, including tremor and difficulties controlling movement. Over a century later, beginning in the 1950s, numerous histopathological and neurological discoveries began to appear.
Currently, Parkinson’s disease is described as a progressive neurodegenerative disease. It presents with specific histological and anatomical abnormalities and distinctive clinical characteristics that allow it to be distinguished from other neurodegenerative illnesses. Despite this knowledge, much of Parkinson’s disease remains a mystery, specifically its etiology and definitive treatment.

Parkinson’s Disease is a neurodegenerative disease
Parkinson’s disease is described as a neurodegenerative disease. As the disease progresses, there is a loss of structure and function of neuron cells. Neurodegenerative diseases are characterized by irreversible damage to neurons, which often results in loss of function.
Changes in the Brain
In Parkinson’s disease, this damage occurs in several parts of the brain, including the locus coeruleus, substantia nigra, raphe nuclei, dorsal nucleus of the vagus nerve, cortical areas, and many others. It has been proposed that the degenerative processes occur first in the dorsal nucleus of the vagus nerve and the olfactory bulb, later in the substantia nigra pars compacta and locus coeruleus, and finally in the cortical regions.
This pattern of degeneration could explain the order of clinical symptoms in patients with Parkinson’s disease. Initially, symptoms are almost exclusively motor, but as the illness progresses there is additional functional decline.
In Parkinson’s disease, the main area of neuronal loss is in the substantia nigra pars compacta, a region involved in the neuronal control of movement through the nigrostriatal pathway. This region synthesizes melanin, which gives it a distinct dark coloring. Therefore, one can observe a loss of pigmentation here when neuronal degeneration occurs. Another substance synthesized in this region is the neurotransmitter dopamine. This neurochemical is highly relevant to the pathophysiology and pharmacological treatment of the disease. Another clinically relevant aspect of the lesion site is its unilateral location. This can result in clinical symptoms that appear laterally in the patient. Finally, it is important to note that initial symptoms start to appear when at least 70 to 80% of the neurons in the substantia nigra are lost.
Histological Changes
Distinctive lesions can be found in histological samples of those with Parkinson’s disease. These lesions include the inclusions and accumulations of protein structures in the nigrostriatal neuron cells. These inclusions are called “Lewy bodies” and are described as spherical and eosinophilic. The protein structures include alpha-synuclein, ubiquitin, and alpha B crystalline. Of the three proteins, alpha-synuclein and ubiquitin are the most relevant in Parkinson’s disease. This is because genetic alterations of these proteins are the cause of several hereditary forms of the disease. Furthermore, through immunohistochemical staining, neuronal samples show that alpha-synuclein is located centrally in the inclusion. Ubiquitin, on the other hand, is found diffusely throughout the inclusion. It is important to note that these inclusions are not exclusive to Parkinson’s disease and that content within the inclusions can vary in other diseases.
Histological samples demonstrate other peculiarities in those with Parkinson’s disease. For example, changes with aging in the neurons of the substantia nigra pars compacta are different in healthy individuals compared to those with Parkinson’s disease. There is a dorsomedial loss of neurons in this region with normal aging. On the other hand, in Parkinson’s disease, the caudal and ventrolateral parts of the substantia nigra pars compacta are affected. Another is the area of the neuron where degeneration processes begin. In Parkinson’s disease, there tend to be initial changes around the neuronal endings, followed by cell body lesions.
Clinical Signs and Symptoms
Apart from the histopathological characteristics, Parkinson’s disease also presents with distinct clinical characteristics:
- Patients tend to be between 50 and 70 years of age when symptoms first appear.
- Parkinson’s disease is slightly more prevalent in men than women (3:2 male to female ratio).
- 95% of patients with Parkinson’s disease are described as sporadic cases, with the absence of a family history of the disease.
Its symptoms are also part of a distinct clinical presentation. Symptoms in Parkinson’s are divided into two groups: motor symptoms and non-motor symptoms. The disease typically begins with ipsilateral motor symptoms, which later develop bilaterally. Non-motor symptoms appear in the final stages of the disease. The cardinal motor symptoms found in Parkinson’s disease are:
- Bradykinesia
- Resting tremor
- Rigidity
- Changes in gait
Other minor motor symptoms include akinesia, hypomimia, micrographia, freezing of gait, and many others. Non-motor symptoms include hyposmia or anosmia, constipation, apathy, fatigue, and cognitive impairments.
Movement Slowing
Bradykinesia, or slowing of movement, is commonly explained by the loss of neurons in the substantia nigra. Motor function is regulated by two pathways that involve the cortex and the basal ganglia. These pathways include the 1) direct pathway and 2) indirect pathway. The direct pathway has an agonist effect on movements while the indirect pathway inhibits movements. Neurons in the substantia nigra have an agonist effect on movements.
Neurons of the substantia nigra innervate other neurons in a structure called the striatum. In the striatum, there are two types of dopamine receptors: D2 (inhibitory receptor) and D1 (excitatory receptor). In areas where D1 is present, this region of the striatum activates the direct pathway. In areas where D2 is present, this region of the striatum activates the indirect pathway. Activation of the dopaminergic neurons in the substantia nigra results in activation of the direct pathway due to activation of the D1 receptors and inactivation of the D2 receptors. Due to the neuronal loss of dopaminergic neurons in the substantia nigra in Parkinson’s disease, activation of this pathway decreases, resulting in movement inhibition and slowing of movements.
Tremor, Rigidity, Changes in Gait, and Non-Motor Symptoms
In contrast to bradykinesia, the pathophysiological basis for the other cardinal symptoms (tremor, rigidity and gait disturbances) are not well understood. There are numerous hypotheses for the causes of each, but further research is necessary. The same can be said for the non-motor symptoms as well. While it’s been hypothesized that non-motor symptoms are due to the degeneration of cortical and limbic structures, more research is necessary.
The Two Sub-Types of Parkinson’s Disease
Given the diversity of symptoms, it has been hypothesized that Parkinson’s disease has two distinct sub-types. The two sub-types are 1) tremor-dominant Parkinson’s disease and 2) postural instability and gait difficulty Parkinson’s disease.
The two types have distinct clinical characteristics. Tremor dominant Parkinson’s disease tends to develop earlier (around 20 – 40 years of age), has a stronger genetic component, and often has a good prognosis. On the other hand, posture instability and gait difficulty Parkinson’s disease typically occurs after the sixth decade of life and also presents with non-motor components.
There are also differences in the histological findings for the two sub-types. Posture instability and gait difficulty Parkinson’s disease tends to show greater neuronal loss and more cortical Lewy bodies. In contrast, tremor dominant Parkinson’s disease often demonstrates lesions in the midbrain, specifically the retrorubral A8 field. However, more research is necessary to support these findings.
Differentiating Parkinson’s Disease from other Disorders
It’s important to understand the histological and anatomical abnormalities, and distinct clinical characteristics, in order to differentiate Parkinson’s disease from other diseases. In particular, it’s important to distinguish Parkinson’s disease from other diseases that cause Parkinsonism and other neurodegenerative disorders.
Parkinsonism
The symptoms commonly found in Parkinson’s disease are often grouped together under the term “Parkinsonism” and can be found secondary to other illnesses. One cause of Parkinsonism is exposure to toxic substances, such as organophosphate pesticides or 1-methyl-1-phenyl-1,2,3,6-tetrahidropiridina. While the signs and symptoms are similar to Parkinson’s disease, a patient’s history will typically reveal exposure to these toxins.
Another environmental cause of Parkinsonism is an infection. Termed post-encephalitic Parkinsonism, this disease was originally observed in the early 1900s. As the name implies, post-encephalitic Parkinsonism also shares clinical similarities with Parkinson’s disease. However, similar to a case of Parkinsonism induced by chemical components, a patient’s history could reveal exposure to this secondary cause.
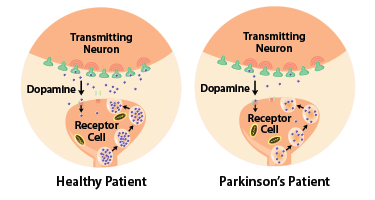
Parkinson’s Disease
Other Neurodegenerative Diseases
Other diseases that mimic Parkinson’s disease are striatonigral degeneration and progressive supranuclear palsy. These three diseases (Parkinson’s disease, striatonigral degeneration, and progressive supranuclear palsy) all have a degenerative process. However, there are differences at the histopathological level.
Striatonigral degeneration differs from Parkinson’s disease because the degeneration process is much more diffuse and lesions in the substantia nigra and the locus coeruleus tend to be less severe. Striatonigral degeneration also has intracellular inclusions of alpha-synuclein but presents with glial cell inclusion as well.
Progressive supranuclear palsy also as a greater diffuse pattern of neuronal cell degeneration. It also presents with severe mesencephalic roof atrophy, resulting in a Mickey Mouse shape due to the prominence of the cerebral peduncles. This disease is also different in terms of cell inclusion content. Progressive supranuclear palsy, instead of accumulating alpha-synuclein, accumulates tau proteins. Clinically, it’s important to look for early imbalance or vertical gaze alterations because these are characteristics of Progressive supranuclear palsy. These appear in later stages or do not appear in Parkinson’s disease.
Other neurodegenerative diseases that should be differentiated from Parkinson’s disease are Alzheimer’s disease, Pick’s disease, and Dementia with Lewy bodies. Alzheimer’s disease can present as Parkinsonism after cognitive changes occur. Cognitive and motor impairments appear inversely when comparing Alzheimer’s disease to Parkinson’s disease, in that Parkinson’s disease begins with motor impairments while Alzheimer’s disease begins with cognitive impairments. This can be associated with the location of the histopathological changes. While Parkinson’s lesions are mainly found in the substantia nigra and the locus coeruleus, in Alzheimer’s disease there is generalized cortical atrophy. Also, the inclusions in Alzheimer’s disease vary because they contain intracellular tau proteins and extracellular beta-amyloid plaques.
The last neurodegenerative disease, dementia with Lewy bodies, can be hard to distinguish from Parkinson’s disease due to their similar histopathology and clinical characteristics. In the histopathological studies, both diseases present with Lewy bodies with alpha-synuclein. However, the location varies between diseases, as dementia with Lewy bodies tends to present in a diffuse manner. Clinically, both diseases present with Parkinsonism, however, the early onset of hallucinations and limbic dysfunction can help differentiate dementia with Lewy bodies. Due to its heterogeneous characteristics, dementia with Lewy bodies has been said to be a Lewy body variant of Alzheimer’s.
Mixed Neurodegenerative Diseases
In order to know what a disease is, a clear definition must be used to distinguish it from other similar diseases. Parkinson’s disease is defined as a non-reversible, progressive neurodegenerative disease that leads to significant motor impairments. However, it can still be difficult to diagnose Parkinson’s disease due to its similarities with other neurodegenerative diseases.
Therefore, the greatest challenge is identifying mixed neurodegenerative diseases. Each neurodegenerative disease has its own particular symptoms that aids in diagnosis, but sometimes two different neurodegenerative diseases can coexist together. For example, the histopathological findings associated with Alzheimer’s disease have been found in 40% of patients diagnosed with Parkinson’s disease. There is no doubt that diagnosing clinically overlapping disorders can be a challenge, even for expert clinicians. Therefore, these complex cases are most often accurately diagnosed only post-mortem.
Parkinson’s disease is a non-reversible, progressive neurodegenerative disease that initially affects the substantia nigra and the locus coeruleus, and later affects other parts of the brain. This results in initial motor impairments followed by impairment of non-motor systems, such as cognitive, emotional, and other neurological functions.
Regardless of the similarities to other neurodegenerative diseases, Parkinson’s disease has a precise clinical definition that facilitates accurate diagnosis. However, further research is needed to discover the pathogenic mechanisms involved. This would greatly benefit the treatment of the disease, as current pharmacological therapy is not curative and only limited improvement of symptoms has been managed. Upon discovery of the precise pathogenic mechanism, it is possible that pharmacological agents could be designed to prevent or even reverse the neurodegeneration that occurs in Parkinson’s disease.
-
 Symptoms and Signs of Parkinson’s disease
Symptoms and Signs of Parkinson’s diseaseThere are several early Signs and Symptoms of Parkinson's Disease. This article explains each of the Parkinson's Disease...
-
Parkinson’s Disease: Testing and Diagnosis
Early diagnosis of Parkinson's Disease is highly essential. Parkinson's Diagnosis and Test is shown in this article. Read...
-
What Causes Parkinson’s Disease?
Want to know what causes Parkinson's Disease? This article lists all the Parkinson's Disease Causes. Read it now!
-
Parkinson’s Disease Treatment Options
A good Parkinson's Disease treatment can help treat or even cure Parkinson's Disease. Read this article to know...
-
9 Early Warning Signs Of Dementia
Dementia is a cluster of neurodegenerative disorders that slowly rob people of their cognitive abilities, memories and emotional...
-
5 Signs and Symptoms of Vitamin E Deficiency
Do you know the signs and symptoms of vitamin E deficiency? Find out if you are getting enough...
-
Multiple Sclerosis Treatment: Types of MS Treatment
A good Multiple Sclerosis treatment can help treat or even cure Multiple Sclerosis. Read this article to know...
-
8 Signs and Symptoms of Celiac Disease
Do you know the signs and symptoms of Celiac disease? Find out in this article and see what...
-
Epilepsy: Types, Symptoms, Causes, Diagnosis, and Treatment
Epilepsy is a chronic condition. Find out its symptoms, causes, treatments, diagnosis, and more in this detailed article.
-
11 Signs & Symptoms of Lyme Disease
Find out the signs and symptoms of Lyme disease today. In this article, we'll talk about the symptoms...










Brittany Heintz, MS, PhD Candidate
Brittany Heintz is a PhD Neuromechanics Candidate and Lecturer at the University of Wisconsin – Milwaukee. Her research examines how the nervous...Read more
Anh Tran, BHSc, MPH
Anh has a Master of Public Health degree from Boston University School of Public Health. In her previous roles, she has had...Read more